Erkrankung
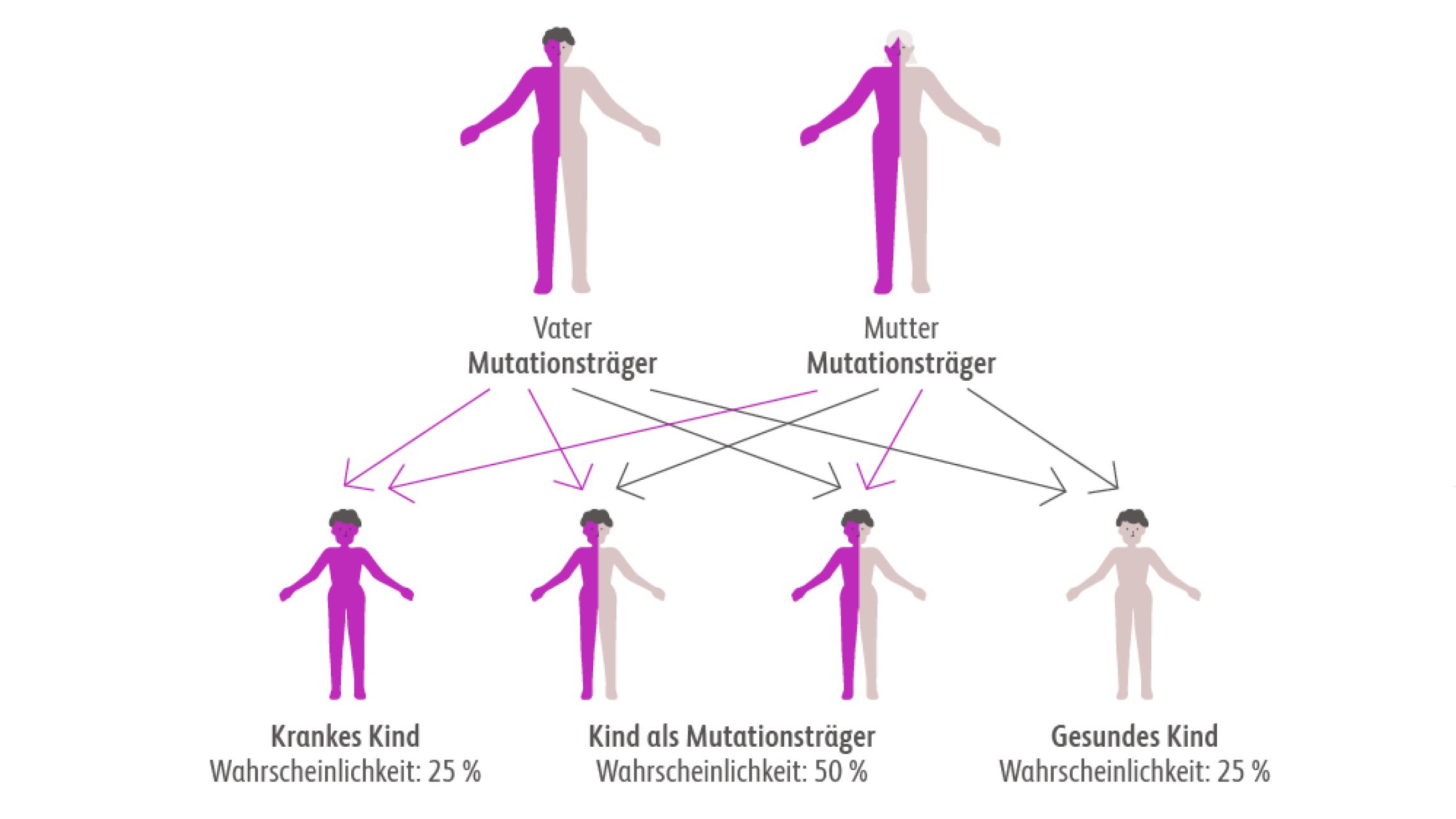
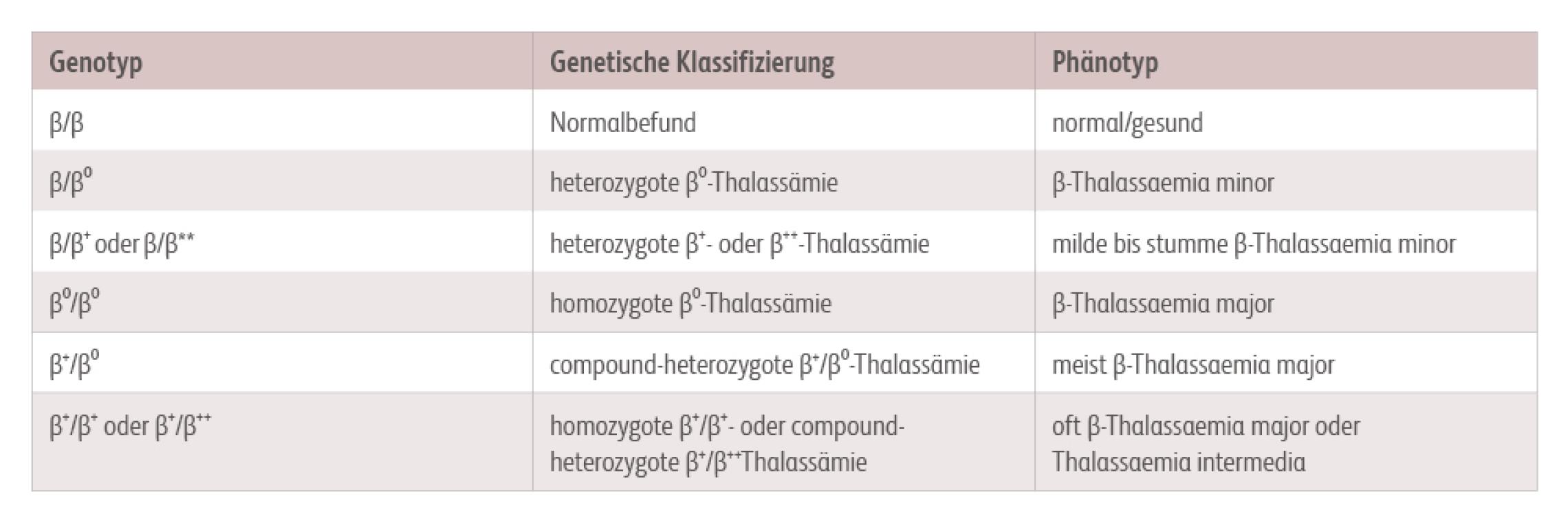
Thalassämien sind Erkrankungen, bei denen die Bildung von Hämoglobin aufgrund einer ganz oder teilweise fehlerhaften Produktion von Globinketten gestört ist. Je nachdem, welche Globinketten betroffen sind, unterscheidet man z. B. die Alpha- und die Beta-Thalassämie. Je nach Schwere der Erkrankung unterscheidet man bei der Beta-Thalassämie zudem eine leichte (minor), mittlere (intermedia) und schwere (major) Form der Erkrankung.ONKOPEDIA Onkopedia Leitlinie. Beta-Thalassämie. Stand Mai 2022. https://www.onkopedia.com/de/onkopedia/guidelines/beta-thalassaemie/@@guideline/html/index.html Abgerufen am 12.06.2024
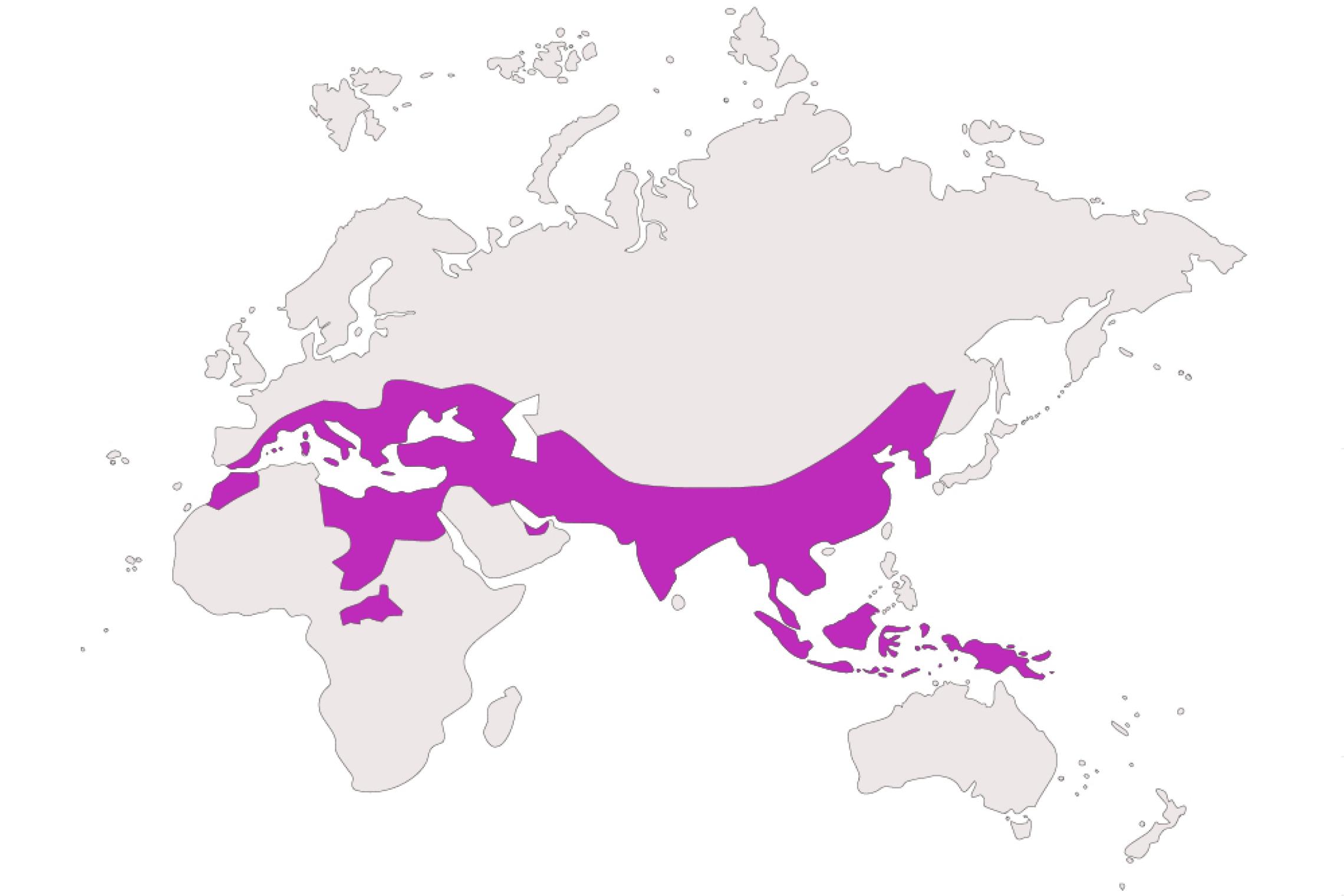
Die Beta-Thalassämie ist weltweit die häufigste erbliche monogenetische Erkrankung. Bei Menschen mit genetischen Wurzeln in Mittelmeerländern, dem Nahen und Mittleren Osten, auf dem indischen Subkontinent, in Südostasien und Afrika ist die Zahl der Anlageträger mit 5 bis 30 % deutlich höher als bei Menschen mit einem anderen genetischen Hintergrund (z. B. 0,01 % bei deutschstämmigen Menschen).THE LANCET, ELSEVIER Taher AT, Weatherall DJ, Cappellini MD: Thalassaemia. Lancet 2018;391:155-167 DOI:10.1016/S0140-6736(17)31822-6 BRITISH JOURNAL PF HAEMATOLOGY, WILEY-BLACKWELL Vetter B, Schwarz C, Kohne E, Kulozik AE. Beta-thalassaemia in the immigrant and non-immigrant German populations. Br J Haematol 1997;97:266-272 PMID:9163586 Als historische Ursache dieser Verteilung gilt, dass leichte Formen einer Thalassämie in Malariaregionen möglicherweise einen gewissen gesundheitlichen Vorteil darstellen.
Aufgrund zunehmender Globalisierung ist auch in Mitteleuropa die Anzahl der Betroffenen in den letzten Jahrzehnten deutlich angestiegen.